FSA is a probabilistic multiple sequence alignment algorithm which uses
a "distance-based" approach to aligning homologous protein, RNA or DNA
sequences. Much as distance-based phylogenetic reconstruction methods
like Neighbor-Joining build a phylogeny using only pairwise divergence
estimates, FSA builds a multiple alignment using only pairwise
estimations of homology. This is made possible by the sequence
annealing technique for constructing a multiple alignment from pairwise
comparisons, developed by Ariel Schwartz in
"Posterior Decoding Methods for Optimization and Control of Multiple
Alignments."
FSA brings the high accuracies previously available only for small-scale analyses of proteins or RNAs
to large-scale problems such as aligning thousands of sequences or megabase-long sequences.
FSA introduces several novel methods for constructing better alignments:
FSA uses machine-learning techniques to estimate gap and
substitution parameters on the fly for each set of input sequences.
This "query-specific learning" alignment method makes FSA very robust: it
can produce superior alignments of sets of homologous sequences
which are subject to very different evolutionary constraints.
FSA is capable of aligning hundreds or even thousands of sequences
using a randomized inference algorithm to reduce the computational
cost of multiple alignment. This randomized inference can be over
ten times faster than a direct approach with little loss of
accuracy.
FSA can quickly align very long sequences using the "anchor
annealing" technique for resolving anchors and projecting them with
transitive anchoring. It then stitches together the alignment
between the anchors using the methods described above.
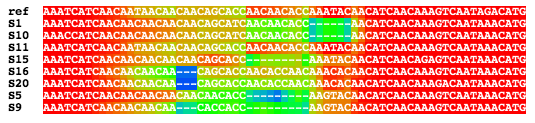
The included GUI, MAD (Multiple Alignment Display), can display
the intermediate alignments produced by FSA, where each character
is colored according to the probability that it is correctly
aligned (see the picture and movie at the top of the page).
FSA is built and installed by running the following commands:
tar xvzf fsa-X.X.X.tar.gz
cd fsa-X.X.X
./configure
make
make install
(Substitute fsa-X.X.X.tar.gz with the name of the file
that you downloaded.)
The FSA executables can then be found in your system's standard
binary directory (e.g., /usr/local/bin). To install to other
locations, see the FAQ.
Alternatively, you may just run FSA from the src/main subdirectory
in which it is built (which does not require running the make
install step)
If you wish to align long sequences, then you must download and install MUMmer,
which FSA calls to get candidate anchors between sequences.
When running ./configure, either have the MUMmer executable in your path
or specify the executable with the --with-mummer option to configure.
See the included README and FAQ
for more information.
Please contact us if you have any build problems.
Webserver
You can submit alignment jobs to the FSA webserver.
Be aware that the webserver may reject alignment jobs which contain many (> 100) sequences
due to computational limitations.
If you wish to align many sequences, then please download and install FSA in order to run the alignment
on your personal computer.
Applications
FSA can be used for all alignment problems, including:
Detailed analysis of a single family of proteins or RNAs.
Large-scale alignment of thousands of sequences (use the --fast option).
Genome alignment of megabases of orthologous sequences.
Citation
Please cite:
Bradley RK, Roberts A, Smoot M, Juvekar S, Do J, Dewey C, Holmes I, Pachter L (2009) Fast Statistical Alignment. PLoS Computational Biology. 5:e1000392.
The FSA manuscript can also be found in the doc/ directory of the FSA source code distribution.
FSA was created by Robert Bradley. It was developed by Robert Bradley,
Colin Dewey, Jaeyoung Do, Sudeep Juvekar, Lior Pachter, Adam Roberts, and
Michael Smoot, along with assistance from many other people.
All have made intellectual contributions and contributed code.
We give our heartfelt thanks to SourceForge for hosting this project.